
藥物選擇性新策略:誘導(dǎo)蛋白蛋白相互作用
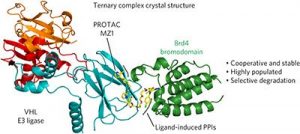
【新聞事件】:這一期的《自然化學(xué)生物學(xué)》雜志發(fā)表了一篇英國鄧迪大學(xué)Ciulli組的一項(xiàng)新工作。這個研究首次得到一個PROTAC化合物MZ1與降解底物BRD4和E3連接酶VHL形成的三組分復(fù)合物的晶體結(jié)構(gòu)。這個晶體結(jié)構(gòu)顯示不僅MZ1同時與這兩個蛋白結(jié)合,還誘導(dǎo)了這兩個蛋白之間的相互作用,形成了一個688平方埃的結(jié)合區(qū)。點(diǎn)變異這個結(jié)合區(qū)的氨基酸雖然不影響MZ1的結(jié)合能,但確實(shí)降低MZ1/Brd4/VHL三組分復(fù)合物的穩(wěn)定性。最后作者根據(jù)這些晶體結(jié)構(gòu)信息設(shè)計(jì)了一個代號為AT1的新PROTAC,可以選擇性增加BRD4(相對于其它BRD)與VHL的結(jié)合能,從而選擇性降解BRD4而沒有影響高度相似的其它BRD。
【藥源解析】:PROTAC是15年前發(fā)明的技術(shù),但近幾年因?yàn)樵趲讉€蛋白尤其是BET族蛋白顯示高細(xì)胞活性和藥物水平的藥代性質(zhì)而成為整個制藥業(yè)關(guān)注的新技術(shù)。和傳統(tǒng)抑制蛋白不同,PROTAC同時與目標(biāo)蛋白和連接酶結(jié)合。后者可以把目標(biāo)蛋白泛素化,從而被蛋白組降解。理論上PROTAC只需要催化劑量即可、也不需要活性很高,因?yàn)槟繕?biāo)蛋白被降解后PROTAC還可以繼續(xù)工作。這可能成為調(diào)控所謂不成藥(undruggable)靶點(diǎn)的一個突破口。
這項(xiàng)工作不僅得到了第一個三組分復(fù)合物的晶體結(jié)構(gòu),還發(fā)現(xiàn)了E3與目標(biāo)蛋白在復(fù)合物中也有結(jié)合、并對復(fù)合物的穩(wěn)定性有重要貢獻(xiàn),這為提高藥物選擇性開辟了一個新路徑。很多蛋白都有多個結(jié)構(gòu)類似、但功能不同甚至相反的同源蛋白。藥物分子有時需要只抑制其中一個,這是小分子藥物發(fā)現(xiàn)的一個重要技術(shù)障礙。現(xiàn)在主要的策略是使用催化腔以外的所謂別構(gòu)抑制劑,但這類抑制劑通常活性欠佳,很多蛋白根本找不到別構(gòu)抑制劑。抗體藥物選擇性好很多但胞內(nèi)靶點(diǎn)多數(shù)無法用抗體控制。
當(dāng)然小分子藥物也可以通過增加與蛋白的結(jié)合面積提高選擇性,但代價是隨著分子量的增加化合物的其它性質(zhì)如藥代性質(zhì)會變差。這篇文章的策略利用了本來就在細(xì)胞內(nèi)存在的配體(即VHL),所以在一定程度上避開了這個問題。當(dāng)然PROTAC分子通常也很大,PK也是一個主要障礙。但現(xiàn)在已有有體內(nèi)活性PROTAC化合物的報道,甚至今年就可能會有進(jìn)入臨床研究的PROTAC藥物。
一個相關(guān)的技術(shù)是Warp Drive Bio的SMART技術(shù)。這個技術(shù)用小分子藥物把內(nèi)源性調(diào)控蛋白(而不是泛素化的E3連接酶)導(dǎo)向到目標(biāo)蛋白,號稱也可以調(diào)控不成藥靶點(diǎn)。當(dāng)然SMART類似傳統(tǒng)抑制劑,要求內(nèi)源性調(diào)控蛋白與目標(biāo)蛋白結(jié)合能足夠高。而PROTAC不需要太高活性。事實(shí)上因?yàn)橐粋€叫做hook effect的現(xiàn)象,PROTAC活性不宜過高,至少與E3和目標(biāo)蛋白的結(jié)合能不能差距太大,否則無法形成所需要的三組分復(fù)合物。如果這種誘導(dǎo)蛋白蛋白相互作用是普遍存在的現(xiàn)象,那么未來藥物的選擇性有望有較大提高。
美中藥源原創(chuàng)文章,轉(zhuǎn)載注明出處并添加超鏈接,商業(yè)用途需經(jīng)書面授權(quán)。
★更多深度解析訪問《美中藥源》~
★ 請關(guān)注《美中藥源》微信公眾號 ★

發(fā)表評論
要發(fā)表評論,您必須先登錄。
藥界的阿波羅11?")
:何為生物類似藥?")
險")
造:恒瑞出售PD-1抗體SHR-1210海外權(quán)益")
:生物類似藥 VS. 化學(xué)仿制藥")

冷淡")
系列談之四:新藥項(xiàng)目是否越早失敗越好?")
- 路人丙: 新藥發(fā)現(xiàn)的低懸果實(shí)
- Pipi_WHU: 新藥發(fā)現(xiàn)的低懸果實(shí)
- 路人丙: Galapagos放棄IPF資產(chǎn)、吉利德空手而歸
- Tan, Chengfang: Galapagos放棄IPF資產(chǎn)、吉利德空手而歸
- 配體效率遭到質(zhì)疑 | 美中藥源: 你笑我無知類藥性本不存在?
- Songdanqing: 你笑我無知類藥性本不存在?
- 路人丙: 中國新藥的新時代
- 康, 榮明: 中國新藥的新時代
合作伙伴









 微信號:美中藥源
微信號:美中藥源